Overfit#6: AlphaFold2: Introduction

The protein folding problem
Proteins are molecular machines that are essential to life. Just to name a few examples: hemoglobin (a protein) carries oxygen in our blood, our immune system fights bacteria using antibodies (proteins), our hair is made of keratine (a protein) ...
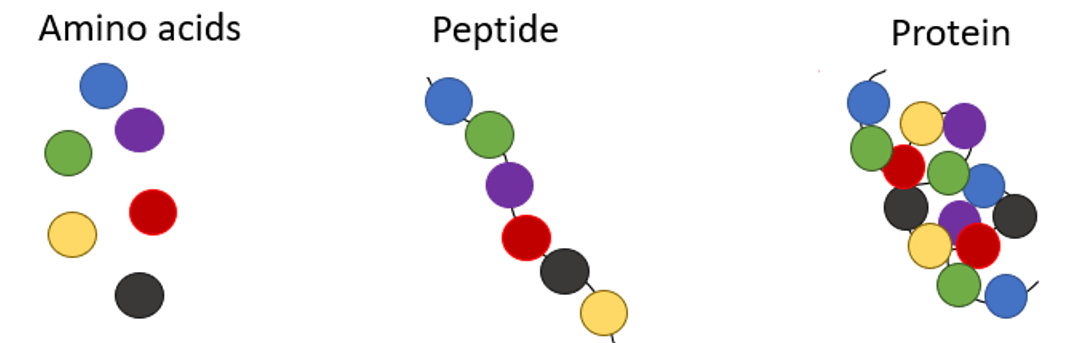
At the atom scale, proteins are linear chains of amino acids, small molecules which act as building blocks. Note that there is a limited number of amino acids (20). That means that with these 20 simple building blocks, you can build any possible protein. Amino acids are often called residues.
🧬 In a nutshell, a protein is a word from a 20-letter alphabet.
Proteins have hydrophilic residues (that like water) and hydrophobic residues (that dislike water). To minimize the contact surface of the latter with water, proteins fold and twist to hide their hydrophobic regions. Thus, the linear chain of amino acids takes a 3D complex structure, a bit like a spaghetti knot.
Why is it so important to know how a protein folds?4
The 3D shape of a protein entirely defines how molecules and other proteins bind to it. In other words, the 3D structure of a protein defines its properties. To quote The New York Times: "If scientists can pinpoint the shape of a particular protein, they can decipher how it operates."
Yet the protein folding problem is hard and expensive to solve. In 2021, the UniProt knowledgebase, a large resource of protein sequences, contained over 60M sequences. For comparison, the Protein Data Bank (PDB), the biggest resource of known protein structures, contained barely over 200K structures.

🤯 With the current techniques, finding the structures of all the known protein sequences would take decades ...
Here is for instance the go-to approach used for most of the structures known to date.

Illustration replicated from the awesome presentation of Simon Kohl, DeepMind [2]
The wet lab approach seems intractable at scale. Could we instead solve this problem computationally?
Simulating proteins with physics-based approaches is out-of-reach for massive use. In fact, one has to compute the dynamics of the protein, and take into account all the interactions between thousands of atoms, at the femtosecond scale. Computational chemists already struggle with small molecules, imagine with entire proteins ...
Now that the plot is in place, let's talk a bit about AI, and discover how Google Deep Mind (almost) cracked this 50-year problem.
CASP14, the break-even
Protein folding is nothing new, scientists have been working on it for decades. Indeed, to accelerate scientific research, the protein folding community even has its biannual competition: the Critical Assessment of Structure Prediction or CASP.
Every two years, the CASP organizers release the sequences of proteins whose 3D structures have been found but not published yet (it acts as a private test dataset). Any researcher is free to submit their structure predictions. The predictions are then compared to the wet lab results to determine which algorithm performs the best.
In 2020, at the 14th edition of CASP (CASP14) a team from Google DeepMind achieved incredible results, beating all their competitors by a MASSIVE margin.
🌟 Their algorithm, AlphaFold21234, is the core topic of this post.

AlphaFold2, a gemstone of engineering
With AlphaFold2, DeepMind introduced an end-to-end model that takes as input a sequence of residues, and returns as output the position in space of each atom of the protein. AlphaFold2 is a sandwich of 2 interleaved modules: the Evoformer and the Structure network, that refine two representations (MSA and pair) of the protein before predicting its structure.
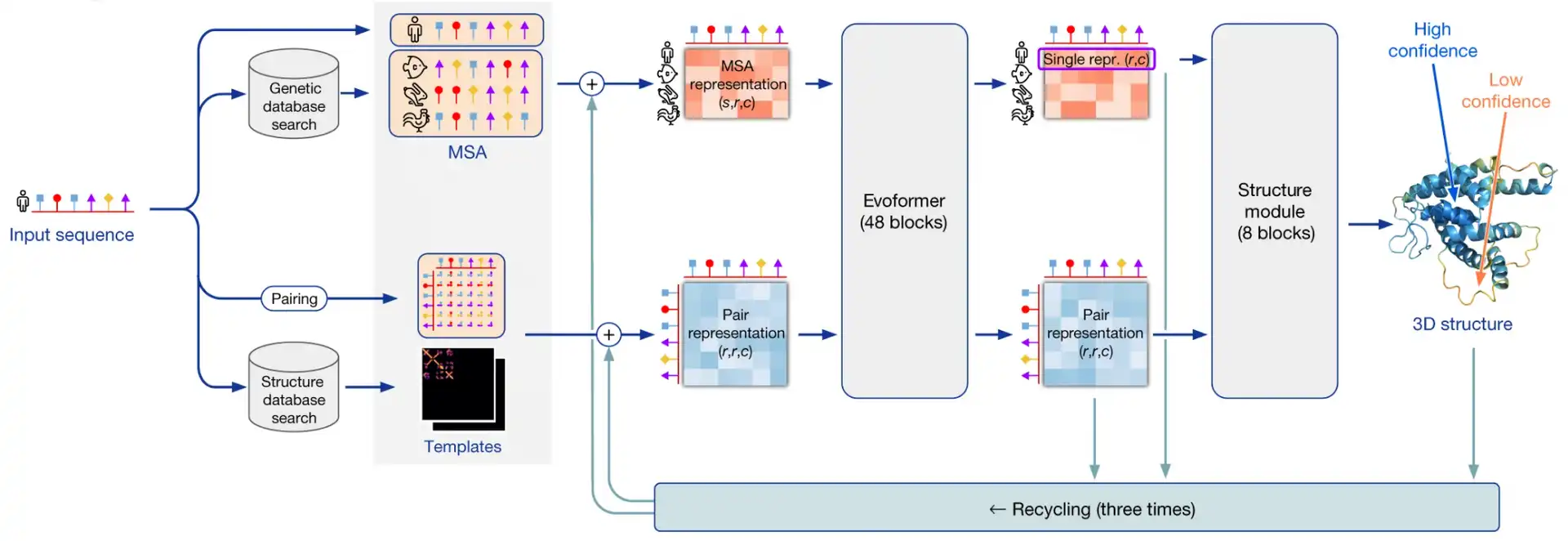
In this series of posts, we will dive together into the architectural and training tricks that make AlphaFold2 such a success. Step by step, we will decipher the secrets of AlphaFold2, a gemstone of engineering.
Table of content
-
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2 ↩
-
Heidelberg AI Talk 5th of May 2022 | Highly Accurate Protein Structure Prediction with AlphaFold | Simon Kohl, DeepMind. https://www.youtube.com/watch?v=tTN0MM2CQLU ↩
-
DeepMind website, AlphaFold technology: https://deepmind.google/technologies/alphafold/ ↩
-
Supplementary Information for: Highly accurate protein structure prediction with AlphaFold: https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-021-03819-2/MediaObjects/41586_2021_3819_MOESM1_ESM.pdf ↩↩